
There are many ways to construct a phylogenetic tree, and after we have done so we are usually expected to indicate something about "branch support", such as bootstrap values or bayesian posterior probabilities. Rarely, however, do people indicate whether there is much tree-like phylogenetic information in their dataset in the first place — it is simply assumed that there must be (fingers crossed, touch wood).
Recently, this latter issue has been addressed for bayesian analysis by:
Paul O. Lewis, Ming-Hui Chen, Lynn Kuo, Louise A. Lewis, Karolina Fučíková, Suman Neupane, Yu-Bo Wang, Daoyuan Shi. (2016) Estimating Bayesian phylogenetic information content. Systematic Biology 65: 1009-1023.They develop a methodology for "measuring information about tree topology using marginal posterior distributions of tree topologies", and apply it to two small empirical datasets. That is, we can now work out something about "[substitution] saturation and detecting conflict among data partitions that can negatively affect analyses of concatenated data."
However, we have long been able to do this with data-display phylogenetic networks. More to the point, we can do it in a second or two, without ever constructing a tree. More pedantically, if the network construction produces a tree, then we know there is tree-like phylogenetic information in the dataset; if we get a network then there is little such information. Equally importantly, the network might tell us something about the patterns of non-tree-likeness, which a single-number measurement cannot.
Let's take the first empirical dataset, as described by the authors:
The five sequences of rpsll composing the data set BLOODROOT [three taxa from the angiosperm family Papaveraceae and two monocots] ... were chosen because they represent a case in which horizontal transfer of half of the gene results in different true tree topologies for the 5′ (219 nucleotide sites) and 3′ (237 nucleotide sites) subsets, which allows investigation of information content estimation in the presence of true conflicting phylogenetic signal. We analyzed each half of the data separately and measured phylogenetic dissonance, which is expected to be high in this case.Here is the NeighborNet based on uncorrected distances. The idea that there is something non-tree-like about Sanguinaria seems hard to avoid. Indeed, the network pattern makes recombination an obvious first choice, with part of the sequence matching the Papaveraceae (on the left) and part matching the monocots (on the right). This recombination may be due to HGT.

Now for the second dataset:
The data set ALGAE comprises chloroplast psaB sequences from 33 taxa of green algae (phylum Chlorophyta, class Chlorophyceae, order Sphaeropleales) ... The alignments of just the psaB gene ... were chosen because of their deep divergence, which invites hasty judgements of saturation, especially of third codon position sites. We analyzed second and third codon position sites separately ... to assess which subset has more phylogenetic information.Here are the two NeighborNets based on uncorrected distances. Once again, it is immediately obvious that the third-codon positions have almost no information at all, even for a network, let alone a tree — the terminal branches do not connect in any coherent way. The second-codon positions do have some information, but it is so contradictory that one could not construct a reliable tree. Saturation of nucleotide substitutions is a likely candidate for this situation; and some correction for this saturation would be needed even to construct a reasonable network from these data.
2nd positions:
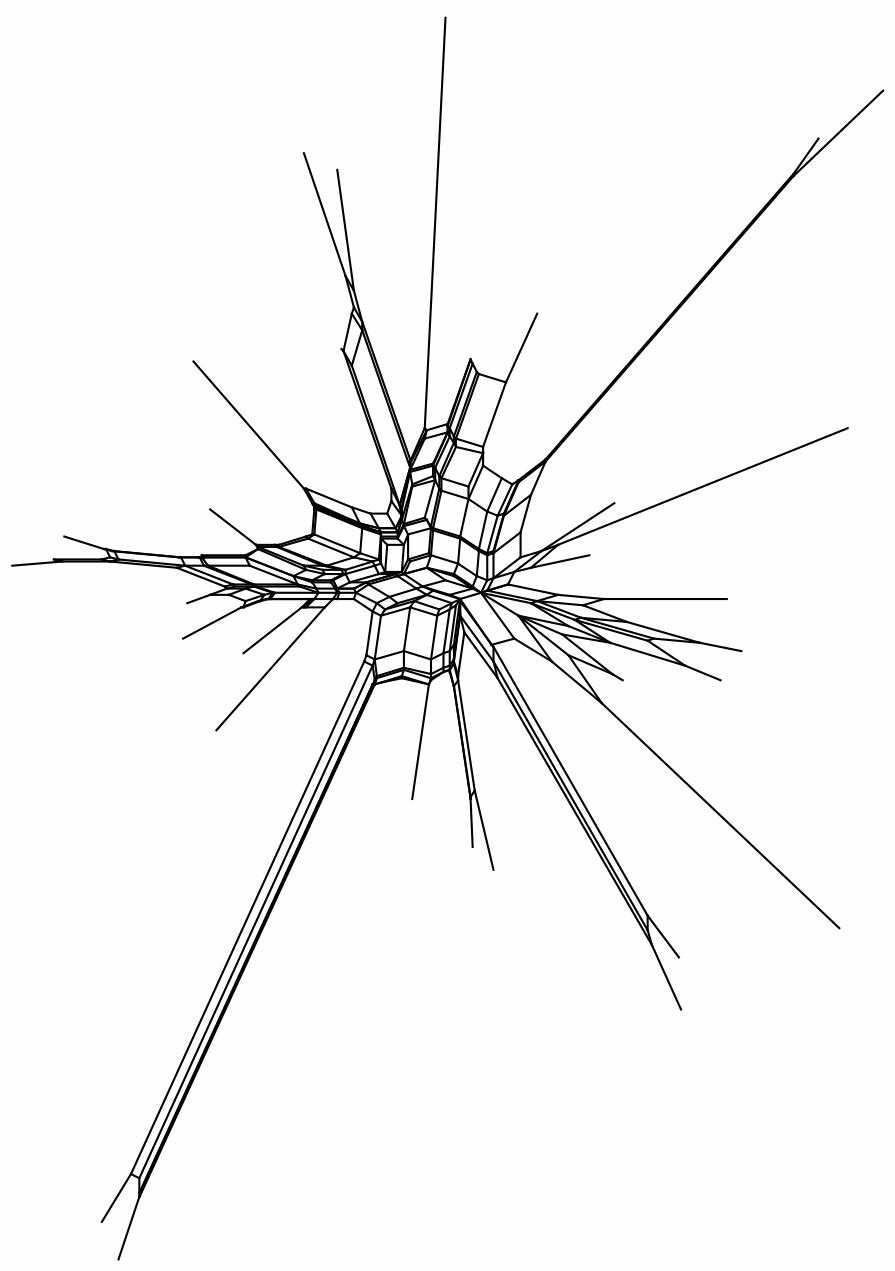
3rd positions:

No comments:
Post a Comment